
Concepto
 Es un grupo de trastornos nerviosos genéticos. También se le puede llamar como atrofia muscular peroneal, neuropatía motora y sensorial hereditaria. Suele comenzar a los 10 ó 20 años, pero a veces lo hace más tarde, hasta los 50-60 años.
Es un grupo de trastornos nerviosos genéticos. También se le puede llamar como atrofia muscular peroneal, neuropatía motora y sensorial hereditaria. Suele comenzar a los 10 ó 20 años, pero a veces lo hace más tarde, hasta los 50-60 años.
Etiología
Desmielización segmentaria crónica de los nervios periféricos, con cambios hipertróficos causados por la remielinación. Geneticamente, el síndrome aparece debido a alteraciones en determinados genes como PMP22, en el 80% de los casos, que se debe a una repetición en tándem del gen dando lugar a una trisomía pricipalmente.
Los nervios periféricos conducen las señales de movimiento y de sensaciones entre el cerebro, la médula espinal y el resto del cuerpo. Los síntomas suelen comenzar durante la adolescencia.
Síntomas
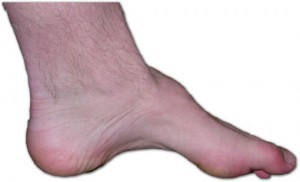
La presentación varía según las distintas familias, pero los individuos afectados de una familia tienden a mostrar una sintomatología similar. Por lo general, comienzo gradual con progresión lenta, deformidad del pie que produce arco alto (cavo) y dedos en martillo, atrofia de las piernas que origina un aspecto de patas de cigüeña, agrandamiento de los nervios, pérdida sensorial u otros signos neurológicos, escoliosis, división de la propiocepción, que muchas veces interfiere en el equilibrio y la marcha, parestesis dolorosas, (en casos avanzados), posible afectación de las manos, ausencia de reflejos tendinosos profundos en muchos pacientes, úlceras de los pies,la cual puede tardar 1 año en volver a regenerarse,debido al debil tejido de la planta del pie, con escasa tendencia a la cicatrización en algunos casos.
Tratamiento
No existe una cura. La enfermedad puede ser tan leve que no perciba que la tiene o ser lo suficientemente severa para debilitarlo. Muchas personas con CMT llevan vidas activas y tienen una vida normal. La fisioterapia, la terapia ocupacional, los aparatos ortopédicos y otros dispositivos y, en algunos casos, la cirugía pueden ayudarlo a lidiar con la enfermedad.